Condición rara que se produce por una lesión del tracto simpático que forma parte del sistema nervioso autónomo, el cual a su vez es el que controla las funciones involuntarias del organismo. Podemos definir este síndrome no como una enfermedad sino como una señal de otro problema médico.
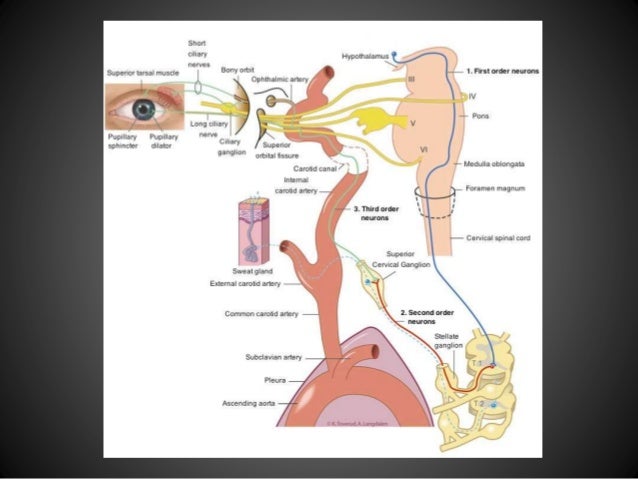
Generalmente se ve afectado un lado del rostro, teniendo como síntoma principal la caída del parpado, disminución de la pupila y de la sudoración del lado del rostro afectado.

Puede ser de origen congénito o adquirido, sin embargo se considera que solo el 5% de los casos son congénitos.

Las causas de padecer este síndrome son muy diversas y entre ellas podemos encontrar:
- Lesiones del tracto simpático.
- Lesiones como tumores o hemorragias cerebrales.
- Placas de esclerosis múltiple en la región del hipotálamo o médula.
- Cirugías del cuello, laringe, tiroides también pueden ser la causa.
- Tumores en la región del cuello, ganglios linfáticos..
Las señales o síntomas generalmente ocurren en un solo lado del rostro y entre ellas se encuentran:
- Caída del parpado superior y ligera elevación del inferior.
- Reducción de la pupila en el ojo afectado.
- Inyección conjuntival (ojo rojo).
En general no existe tratamiento para este síndrome, este va a depender exclusivamente de la causa del mismo. Muchas veces desaparece cuando se erradica la enfermedad que lo produce.
Por último y como curiosidad deciros que es un síndrome secundario a la anestesia epidural debido a un bloqueo de las fibras simpáticas del ganglio estrellado. Esta complicación se produce por la migración no esperada del analgésico local administrado en el espacio epidural o dentro del paquete aponeurótico vasculonervioso en un bloqueo del plexo braquial.

Es un cuadro de rápida evolución, benigno, que desaparece en horas, sin dejar secuelas y que puede ocurrir en pacientes sin ningún antecedente patológico.