El síndrome DIRA consiste en la deficiencia del antagonista del receptor de la IL-1, es una enfermedad genética rara. Los niños que la padecen presentan inflamaciones cutáneas y óseas graves. Otros órganos, como los pulmones también pueden verse afectados.
Si esta enfermedad no se trata a tiempo, la enfermedad puede llevar a una discapacidad importante o incluso causar la muerte.
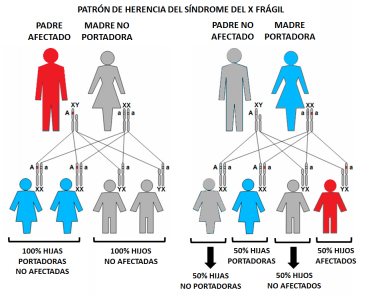
El síndrome DIRA es muy poco frecuente y como comenté anterior mente es una enfermedad de causa genética, el gen responsable se lama IL1RN. Este gen produce una proteína que juega un papel muy importante en la resolución natural de la inflamación. Es decir, si este gen sufre una mutación el paciente desarrolla una inflamación constante.
Los principales síntomas de la enfermedad son la inflamación cutánea y la inflamación ósea. La cutánea se caracteriza por enrojecimiento, descamación y formación de pústulas, Aparece de forma espontánea y en cualquier parte del cuerpo.
Por otra parte la ósea, se caracteriza por inflamaciones dolorosas, a menudo con enrojecimiento y calor en la piel de la zona que cubre al hueso. Normalmente la inflamación implica el periostio, por lo tanto los niños afectados suelen estar irritables y abatidos. Esto puede provocar inapetencia y retraso en el crecimiento.
Cabe añadir que el Síndrome DIRA únicamente se ha reconocido en niños, por consiguiente se desconocen los síntomas en la edad adulta.
La enfermedad no se puede curar ni se puede controlar adecuadamente con antiinflamatorios pero si se puede controlar parcialmente con ayuda de dosis altas de coticoesteroides. Anakinra es la forma de IL1RA producida de forma artificial actualmente y es del único modo que se puede mantener la enfermad bajo control. Es un medicamento que el paciente deberá de usar toda su vida, ya que solo así los síntomas desaparecerarn parcialmente.
Por último avisar que si el paciente dejase de medicarse , la enfermedad volvería a aparecer y con ella los síntomas que son potencialmente mortales.

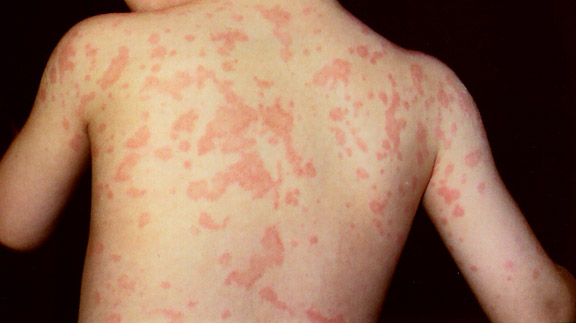 Uno de los principales signos de este síndrome es la presencia de una erupción en la infancia o la primera infancia, a menudo alrededor de los 4 meses de edad. La artritis es el encontrar más común de los pacientes de Blau, y comienza en el plazo de los primeros 10 años de vida.
Uno de los principales signos de este síndrome es la presencia de una erupción en la infancia o la primera infancia, a menudo alrededor de los 4 meses de edad. La artritis es el encontrar más común de los pacientes de Blau, y comienza en el plazo de los primeros 10 años de vida.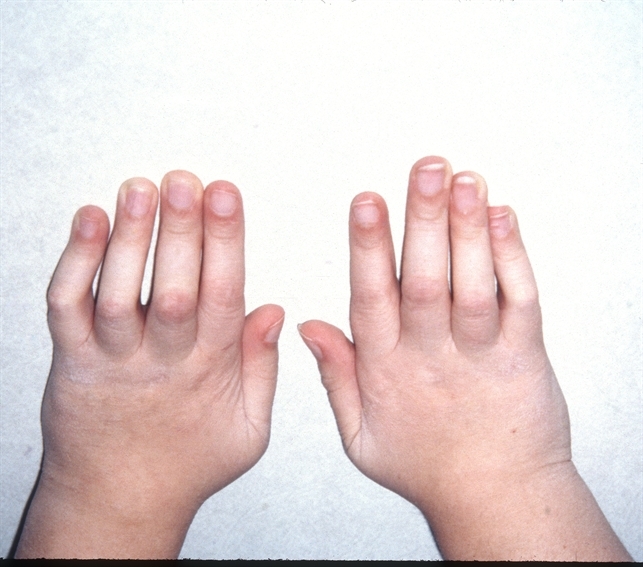 Están también presentes granulomas, quistes sinoviales.
Están también presentes granulomas, quistes sinoviales.